Фармакодинамические свойства
Механизм действия
Алектиниб является высокоселективным и мощным ингибитором ALK и RET тирозинкиназы. В доклинических исследованиях ингибирование активности тирозинкиназы ALK приводило к блокированию нисходящих сигнальных путей, в том числе STAT 3 и PI3K/AKT, а также индукции гибели опухолевых клеток (апоптоз).
Алектиниб продемонстрировал активность в условиях in vitro и in vivo в отношении мутантных форм фермента ALK, включая мутации, ответственные за резистентность к кризотинибу. Основной метаболит алектиниба (М4) продемонстрировал схожую активность в условиях in vitro.
По данным доклинических исследований, алектиниб не является субстратом Р-гликопротеина или белка резистентности рака молочной железы (BCRP), являющихся эффлюксными транспортерами через гематоэнцефалический барьер и, следовательно, способных поступать и распределяться в центральной нервной системе.
Клиническая эффективность и безопасность
ALK-положительный немелкоклеточный рак легких
Пациенты, ранее не получавшие лечения
Безопасность и эффективность препарата Алеценза изучалась в глобальном рандомизированном открытом клиническом исследовании фазы III (ВО28984, ALEX) с участием пациентов с ALK- положительным немелкоклеточным раком легких, ранее не получавших лечения. Перед рандомизацией в исследование требовалось проведение централизованного тестирования на определение положительного статуса экспрессии белка ALK. в образцах тканей, взятых у пациентов, методом иммуногистохимического (ИГХ) анализа Ventana anti-ALK (D5F3).
В исследование фазы III всего было включено 303 пациента, 151 из которых были рандомизированы в группу кризотиниба и 152 – в группу Алеценза, где пациенты получали препарат Алеценза перорально в рекомендуемой дозе по 600 мг два раза в сутки.
Стратификационными факторами при рандомизации являлись оценка по шкале ECOG PS (0/1 или 2), раса (азиатская или неазиатская) и наличие метастазов ЦНС на момент включения в исследование (да или нет). Первичной конечной точкой исследования являлось подтверждение большей эффективности препарата Алеценза по сравнению с кризотинибом по результатам выживаемости без прогрессирования заболевания (PFS) по оценке исследователя при помощи Критериев оценки ответа при солидных опухолях (RECIST 1.1). Исходные демографические характеристики и характеристики заболевания в группе приема препарата Алеценза: медиана возраста 58 лет (54 года в группе кризотиниба), 55% пациентов были женского пола (58% в группе кризотиниба), 55% пациентов неазиатской расы (54% в группе кризотиниба), 61% некурящие (65% в группе кризотиниба), у 93% пациентов оценка по шкале ECOG PS была от 0 до 1 (93% в группе кризотиниба), 97% пациентов имели IV стадию заболевания (96% в группе кризотиниба), у 90% пациентов была гистологически подтвержденная аденокарцинома (94% в группе кризотиниба), у 40% были метастазы в ЦНС на момент включения в исследование (38% в группе кризотиниба), у 17% ранее получали облучению ЦНС (14% в группе кризотиниба).
По результатам первичного анализа исследование достигло первичной конечной точки со статистически достоверным улучшением по выживаемости без прогрессирования заболевания по оценке исследователя. Данные по эффективности обобщены в таблице 4; на рисунке 1 представлена кривая Каплана-Мейера, демонстрирующая результаты по выживаемости без прогрессирования заболевания по оценке исследователя.
Таблица 4 Обзор результатов по эффективности из исследования ВО28984 (ALEX)
|
|
Кризотиниб
N=151
|
Алеценза
N=152
|
|
Медиана продолжительности
периода наблюдения (месяцы)
|
17.6
(диапазон 0.3-27.0)
|
18.6
(диапазон 0.5-29.0)
|
|
Первичный показатель
эффективности
|
|
|
|
Выживаемость без
прогрессирования заболевания, PFS
(иссл.)
|
|
|
Количество пациентов с
явлениями, n (%)
Медиана (месяцы)
[95% ДИ]
|
102 (68%)
11.1
[9.1; 13.1]
|
62 (41%)
н.м.б.о.
[17.7; н.м.б.о.]
|
|
|
|
|
Отношение рисков
[95% ДИ]
Стратифицированное логранговое
р-значение
|
0.47
[0.34; 0.65]
р <0. 0001
|
|
|
|
|
|
Вторичные показатели
эффективности
|
|
|
|
|
|
|
|
Выживаемость без
прогрессирования заболевания, PFS
(ННК)
|
|
|
Количество пациентов с
явлениями, n
(%)
Медиана (месяцы)
[95% ДИ]
|
92 (61%)
10.4
[7.7; 14.6]
|
63 (41%)
25.7
[19.9; н.м.б.о.]
|
|
|
|
|
Отношение рисков (HR)
[95% ДИ]
Стратифицированное логранговое
р-значение
|
0.50
[0.36; 0.70]
р <0.0001
|
|
Время до прогрессирования в ЦНС
(ННК)*, **
|
|
|
Количество пациентов с
явлениями, n (%)
|
68 (45%)
|
18 (12%)
|
|
|
|
|
Отношение рисков (HR) в зависимости от причины
[95% ДИ]
Стратифицированное логранговое
р-значение
|
0.16
[0.10; 0.28]
p<0/0001
|
|
|
|
|
Совокупная частота случаев
прогрессирования в ЦНС за 12 месяцев (ННК)
[95% ДИ]
|
41.4%
[33.2; 49.4]
|
9.4%
[5.4; 14.7]
|
|
Частота объективного ответа (ORR) (иссл.)*, ***
|
|
|
|
Количество пациентов, достигших
ответа, n (%) [95% ДИ]
|
114 (75.5%)
[67.8; 82.1]
|
126 (82.9%)
[76.0; 88.5]
|
|
Общая выживаемость*
|
|
|
Количество пациентов с
явлениями, n (%)
Медиана (месяцы)
[95% ДИ]
|
40 (27%)
н.м.б.о.
[н.м.б.о.]
|
35 (23%)
н.м.б.о.
[н.м.б.о.]
|
|
Отношение рисков (HR) [95% ДИ]
|
0.76
[0.48; 1.20]
|
|
Продолжительность ответа (иссл.)
|
N=114
|
N=126
|
Медиана (месяцы)
[95% ДИ]
|
11.1
[7.9; 13.0]
|
н.м.б.о.
[н.м.б.о.]
|
|
Частота объективного ответа (CNS-ORR) у пациентов с измеримыми метастазами в ЦНС на момент включения в
исследование
|
N=22
|
N=21
|
Количество пациентов, достигших
ответа, n (%)
[95% ДИ]
|
11 (50.0%)
[28.2; 71.8]
|
17 (81.0%)
[58.1; 94.6]
|
|
Полный ответ (CNS-CR), n
(%)
|
1 (5%)
|
8 (38%)
|
|
|
|
|
|
Продолжительность ответа (CNS-DOR), медиана (месяцы)
|
5.5
|
17.3
|
|
[95% ДИ]
|
[2.1; 17.3]
|
[14.8; н.м.б.о.]
|
|
Частота объективного ответа (CNS-ORR) у пациентов с измеримыми и неизмеримыми метастазами в ЦНС на
момент включения в исследование (ННК)
|
N=58
|
N=64
|
Количество пациентов, достигших ответа,
n (%)
[95% ДИ]
|
15 (25.9%)
[15.3; 39.0]
|
38 (59.4%)
[46.4; 71.5]
|
|
|
|
|
|
Полный ответ (CNS-CR), n
(%)
|
5 (9%)
|
н.м.б.о.
|
|
|
|
|
|
Продолжительность ответа (CNS-DOR), медиана (месяцы)
|
3.7
|
н.м.б.о.
|
|
[95% ДИ]
|
[3.2; 6.8]
|
[17.3; н.м.б.о.]
|
* Ключевые вторичные конечные точки в рамках многоуровневого анализа
** Анализ конкурирующих рисков прогрессирования в ЦНС, системного прогрессирования и смерти в качестве конкурирующих явлений
*** 2 пациента в группе кризотиниба и 6 пациентов в группе приема алектиниба достигли полного ответа
ДИ = доверительный интервал; ЦНС = центральная нервная система; CR = полный ответ; DOR = продолжительность ответа; HR = отношение рисков; ННК = независимый наблюдательный комитет; иссл. = исследователь; н.м.б.о. = не может быть оценено; ORR = частота объективного ответа; PFS = выживаемость без прогрессирования
У пациентов, имеющих метастазы в ЦНС на момент включения в исследование (отношение рисков HR = 0,40, 95% ДИ: 0,25-0,64; медиана выживания без прогрессирования заболевания в группе Алеценза = н.м.б.о., 95% ДИ: 9,2-н.м.б.о., медиана выживания без прогрессирования заболевания в группе кризотиниба = 7,4 месяца, 95% ДИ: 6,6-9,6) и не имеющих метастазы в ЦНС на момент включения в исследование (отношение рисков HR = 0,51, 95% ДИ: 0,33-0,80, медиана выживания без прогрессирования заболевания в группе Алеценза = н.м.б.о., 95% ДИ: н.м.б.о.-н.м.б.о., медиана выживания без прогрессирования заболевания в группе кризотиниба = 14,8 месяцев, 95% ДИ: 10,8- 20,3) показатель выживаемости без прогрессирования заболевания был выше, что указывает на более высокую эффективность препарата Алеценза по сравнению с кризотинибом в обеих подгруппах.
Рисунок 1: Кривая Каплана-Мейера для выживаемости без прогрессирования заболевания по оценке исследователя в исследовании ВО28984 (ALEX)
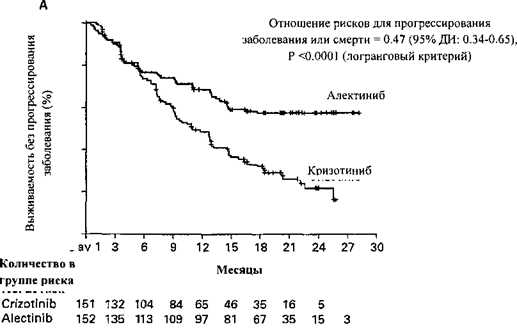 Пациенты, получавшие лечение кризотинибом
Пациенты, получавшие лечение кризотинибом
Безопасность и эффективность препарата Алеценза у пациентов с ALK-положительным НМРЛ, ранее получавших кризотиниб, исследовались в двух клинических исследованиях фазы I/II (NP28673 и NP28761).
NP28673
Исследование NP28673 являлось многоцентровым исследованием фазы I/II с одной группой лечения, проводившееся с участием пациентов с прогрессирующим ALK-положительным НМРЛ, у которых заболевание прогрессировало в ходе лечения кризотинибом. Помимо кризотиниба, пациенты могли проходить химиотерапию. Всего 138 пациентов были включены в фазу II исследования, в ходе которой они получали препарат Алеценза внутрь в рекомендуемой дозе 600 мг два раза в сутки.
Первичной конечной точкой являлась оценка эффективности препарата Алеценза на основании частоты объективного ответа (ORR) согласно централизованной оценке независимого наблюдательного комитета при помощи Критериев оценки ответа при солидных опухолях (RECIST 1.1) у пациентов, ранее проходивших цитотоксическую химиотерапию. При снижении доверительного интервала для оцененной частоты объективного ответа выше предварительно установленного предела 35% достигается статистически достоверный результат.
Демографические характеристики пациентов соответствовали популяции пациентов с ALK- положительным НМРЛ. Общие демографические характеристики в исследовании были следующими: 67% – европеоидная раса, 26% – монголоидная раса, 56% – женщины, медиана возраста – 52 года. Большинство пациентов ранее не курили (70%). На момент включения в исследование статус по шкале ECOG (Восточная объединенная группа онкологов) от 0 до 1 был зарегистрирован у 90,6% пациентов, а 2 – у 9,4% пациентов. На момент включения в исследование 99% пациентов имели IV стадию заболевания, у 61% были метастазы в головной мозг, а у 96% пациентов опухоли были классифицированы как аденокарцинома. Среди пациентов, включенных в исследование, у 20% наблюдалось значительное прогрессирование в ходе терапии кризотинибом, а у 80% заболевание ранее прогрессировало в ходе лечения кризотинибом и не менее одного курса химиотерапии.
NP28671
Исследование NP28671 являлось многоцентровым исследованием фазы I/II с одной группой, проводившимся с участием пациентов с ALK-положительным НМРЛ, у которых заболевание ранее прогрессировало в ходе лечения кризотинибом. Помимо кризотиниба, пациенты могли проходить химиотерапию. Всего 87 пациентов были включены в фазу II исследования, в ходе которой они получали препарат Алеценза внутрь в рекомендуемой дозе 600 мг два раза в сутки.
Первичной конечной точкой являлась оценка эффективности препарата Алеценза на основании частоты объективного ответа (ORR) согласно централизованной оценке независимого наблюдательного комитета при помощи Критериев оценки ответа при солидных опухолях (RECIST 1.1). При снижении доверительного интервала для оцененной частоты объективного ответа выше предварительно установленного предела 35% достигается статистически достоверный результат.
Демографические характеристики пациентов соответствовали популяции пациентов с ALK-положительным НМРЛ. Общие демографические характеристики в исследовании были следующими: 84% – европеоидная раса, 8% – монголоидная раса, 55% – женщины. Медиана возраста составила 54 года. Большинство пациентов ранее не курили (62%). На момент включения в исследование статус по шкале ECOG (Восточная объединенная группа онкологов) от 0 до 1 был зарегистрирован у 89,7% пациентов, а 2 – у 10,3% пациентов. На момент включения в исследование 99% пациентов имели IV стадию заболевания, у 60% были метастазы в головной мозг, а у 94% пациентов опухоли были классифицированы как аденокарцинома. Среди пациентов, включенных в исследование, у 26% наблюдалось значительное прогрессирование в ходе терапии кризотинибом, а у 74% заболевание ранее прогрессировало в ходе лечения кризотинибом и не менее одного курса химиотерапии.
Основные результаты по эффективности из исследований NP28673 и NP28671 обобщены в таблице 5. Обзор анализа объединенных данных по конечным точкам ЦНС представлен в таблице 6.
Таблица 5 Результаты по эффективности из исследований NP28673 и NP28671
|
|
NP28673
Алеценза 600 мг два раза в сутки
|
NP28671
Алеценза 600 мг два раза в сутки
|
|
Медиана продолжительности
периода наблюдения (месяцы)
|
21
(диапазон 1-30)
|
17
(диапазон 1-29)
|
|
Первичный показатель эффективности
|
|
|
|
Частота объективного ответа (ORR) в популяции RE
|
N=122а
|
N=67b
|
Количество пациентов, N (%)
[95% ДИ]
|
62 (50.8%)
[41.6%, 60.0%]
|
35 (52.2%)
[39.7%, 64.6%]
|
|
|
|
|
|
Частота объективного ответа (ORR) у пациентов, ранее проходивших
химиотерапию
|
N=96
|
|
Количество пациентов, N (%)
[95% ДИ]
|
43 (44.8%)
[34.6%, 55.3%]
|
|
|
Вторичные показатели
эффективности
|
|
|
|
Продолжительность ответа (DOR) (ННК)
|
N=62
|
N=35
|
|
Количество пациентов с
явлениями, N (%)
|
36 (58.1%)
|
20 (57.1%)
|
|
Медиана (месяцы)
|
15.2
|
14.9
|
|
[95% ДИ]
|
[11.2, 24.9]
|
[6.9, н.м.б.о.]
|
|
|
|
|
|
Выживаемость без
прогрессирования заболевания (PFS) (ННК)
|
N=138
|
N=87
|
|
Количество пациентов с
явлениями, N (%)
|
98 (71.0%)
|
58 (66.7%)
|
|
Медиана (месяцы)
|
8.9
|
8.2
|
|
[95% ДИ]
|
[5.6, 12.8]
|
[6.3, 12.6]
|
ДИ = доверительный интервал; DOR = продолжительность ответа; ННК = независимый наблюдательный комитет; н.м.б.о. = не может быть оценено; ORR = частота объективного ответа; PFS = выживаемость без прогрессирования заболевания; RE = с ответом, который может быть оценен
а У 16 пациентов болезнь на момент включения в исследование была неизмерима согласно ННК, поэтому не была включена популяцию с ответом, который может быть оценен.
b У 20 пациентов болезнь на момент включения в исследование была неизмерима согласно ННК, поэтому не была включена популяцию с ответом, который может быть оценен.В исследованиях NP28673 и NP28671 результаты по частоте объективного ответа были однородными по всем подгруппам исходных характеристик пациентов, таких как возраст, пол, раса, статус по шкале ECOG, наличие метастазов в ЦНС и прохождение химиотерапии, особенно если учитывать небольшое число пациентов в некоторых подгруппах.
Таблица 6. Обзор анализа объединенных данных по конечным точкам ЦНС из исследований NP28673 и NP28671
|
Показатели
ЦНС (NP28673 и NP28671)
|
Алеценза 600 мг два раза в сутки
|
|
Пациенты с измеримыми
поражениями ЦНС на момент включения в исследование
|
N=50
|
|
Частота объективного ответа для
ЦНС (CNS
ORR) (ННК)
|
|
|
Количество пациентов, достигших
ответа (%)
|
32 (64.0%)
|
|
[95% ДИ]
|
[49.2%, 77.1%]
|
|
Полный ответ
|
11 (22.0%)
|
|
Частичный ответ
|
21 (42.0%)
|
|
|
|
|
Продолжительность ответа для ЦНС
(CNS
DOR) (ННК)
|
N=32
|
|
Количество пациентов с явлениями
(%)
|
18(56.3%)
|
|
Медиана (месяцы)
|
11.1
|
|
[95% ДИ]
|
[7.6, н.м.б.о.]
|
ДИ = доверительный интервал; DOR = продолжительность ответа; ННК = независимый наблюдательный комитет; ORR - частота объективного ответа; NE = не может быть оцененоДети
Европейское агентство по лекарственным средствам отказалось от обязательства по предоставлению результатов исследований препарата Алеценза по всем подгруппам детской популяции пациентов с карциномой легких (мелкоклеточным и немелкоклеточным раком легких) (сведения о применении у детей см. в разделе «Режим дозирования и способ применения»).
Фармакокинетические свойства
Была дана характеристика фармакокинетических показателей алектиниба и его основного метаболита (М4) у пациентов с ALK-положительным НМРЛ и у здоровых участников. По результатам популяционного фармакокинетического анализа, среднее геометрическое значение (коэффициент вариации, %) равновесных С
max, С
min и AUC
0-12ч алектиниба составляло соответственно приблизительно 665 нг/мл (44,3%), 572 нг/мл (47,8%) и 7430 нг*ч/мл (45,7%). среднее геометрическое значение (коэффициент вариации, %) равновесных С
max, С
min и AUC
0-12ч метаболита М4 составляло соответственно приблизительно 246 нг/мл (45,4%), 222 нг/мл (46,6%) и 2810 нг*ч/мл (45,9%).
Абсорбция
После приема внутрь в дозе 600 мг два раза в сутки после еды у пациентов с ALK-положительным НМРЛ алектиниб всасывался с достижением Т
max через приблизительно 4-6 часов.
Равновесная концентрация алектиниба достигалась через 7 дней при непрерывном приеме в дозе 600 мг два раза в сутки. Коэффициент накопления при приеме в дозе 600 мг два раза в сутки составлял приблизительно 6. Популяционный фармакокинетический анализ подтверждает пропорциональность доз для всего диапазона доз от 300 до 900 мг при приеме после еды.
Абсолютная биодоступность алектиниба в капсулах у здоровых участников составляла 36,9% (90% ДИ: 33,9%, 40,3%) при приеме после еды.
После однократного приема внутрь в дозе 600 мг вместе с жирной высококалорийной пищей экспозиция алектиниба и метаболита М4 повышалась приблизительно в 3 по сравнению с приемом натощак (см. раздел «Режим дозирования и способ применения»).
Распределение
Алектиниб и его основной метаболит М4 активно связываются с белками плазмы человека (>99%) независимо от концентрации действующего вещества. Среднее соотношение концентраций алектиниба и М4 в крови и плазме in vitro соответственно составило 2,64 и 2,50 при приеме в клинически значимых дозах.
Среднее геометрическое значение объема распределения в равновесном состоянии (V
ss) алектиниб после внутривенного введения составило 475 л, что свидетельствует о масштабном распределении в ткани.
По данным in vitro, алектиниб не является субстратом Р-гликопротеина. Алектиниб и метаболит М4 не являются субстратами белка резистентности рака молочной железы (BCRP) и полипептида транспортера органических анионов (ОАТР) 1В1/ВЗ.
Биотрансформация
Исследования метаболизма, проведенные в условиях in vitro, продемонстрировали, что CYP3A4 является основным ферментом, опосредующий метаболизм алектиниба и его основного метаболита М4. По оценкам, 40-50% метаболизма алектиниба приходится на долю данного фермента. По результатам исследования баланса масс у человека было продемонстрировано, что алектиниб и М4 являются основными компонентами, циркулирующими в плазме, и составляют 76% от всей радиоактивной метки в плазме. Среднее геометрическое значение соотношения метаболит/материнское соединение в равновесном состоянии составляет 0,399.
В условиях in vitro и в плазме крови здоровых участников был обнаружен второстепенный метаболит М1b. Вероятно, образование метаболита М1b катализируется комбинацией изоферментов CYP (включая другие изоферменты, помимо CYP3A) и ферментами альдегиддегидрогеназы (ALDH).
Исследования in vitro указывают на то, что ни алектиниб, ни его основной метаболит (М4) не ингибируют CYP1A2, CYP2B6, CYP2C9, CYP2C19 и CYP2D6 при клинически значимых концентрациях. Алектиниб не ингибирует ОАТР1В1/ОАТР1ВЗ, ОАТ1, ОАТЗ и ОСТ2 при клинически значимых концентрациях in vitro.
Элиминация
После однократного приема внутрь алектиниба, меченого
14С, у здоровых участников основная часть меченого
14С выводилась с калом (средний коэффициент извлечения 97,8%) с минимальной долей, выведенной с мочой (средний коэффициент извлечения 0,46%). С калом 84% и 5,8% дозы выводилось соответственно в виде неизмененного алектиниба или метаболита М4.
По результатам популяционного фармакокинетического анализа, кажущийся клиренс алектиниба (CL/F) составил 81,9 л/ч. Среднее геометрическое отдельных значений полувыведения алектиниба составило 32,5 ч. Соответствующие значения для метаболита М4 составили соответственно 217 л/ч и 30,7 ч.
Фармакокинетические характеристики у отдельных категорий пациентов
Почечная недостаточность
Незначительное количество алектиниба и активного метаболита М4 выводятся в неизмененном виде с мочой (<0,2% от дозы). По результатам популяционного фармакокинетического анализа, экспозиция алектиниба и метаболита М4 была схожей у пациентов с легкой и умеренной степенью почечной недостаточности и нормальной функцией почек. Фармакокинетические свойства алектиниба не изучались у пациентов с тяжелой почечной недостаточностью.
Печеночная недостаточность
Элиминация алектиниба происходит главным образом через печень, в связи с чем печеночная недостаточность может повысить концентрацию алектиниба и/или его основного метаболита в плазме. По результатам популяционного фармакокинетического анализа, экспозиция алектиниба и М4 были схожими у пациентов с умеренной печеночной недостаточностью и нормальной функцией печени.
Фармакокинетические свойства алектиниба не изучались у пациентов с тяжелой печеночной недостаточностью.
Влияние возраста, массы тела, расы и пола
Возраст, масса тела, раса и пол не оказывали клинически значимого влияния на системное воздействие алектиниба и метаболита М4. В клинические исследования были включены пациенты с массой тела в диапазоне от 36,9 до 123 кг. Данные о пациентах с крайне высокой массой тела (более 130 кг) отсутствуют (см. раздел «Режим дозирования и способ применения»).
Данные доклинической безопасности
Канцерогенность
Исследований канцерогенности по установлению канцерогенного потенциала препарата Алеценза не проводилось.
Мутагенность
Алектиниб не проявлял мутагенной активности in vitro в ходе испытания на обратные мутации у бактерий (тест Эймса), однако, он вызывал незначительное увеличение численных аберраций в цитогенетическом анализе in vitro, проводившемся на клетках легких китайских хомячков с метаболической активацией, а также в микроядрах спинного мозга крыс в микроядерном тесте. Механизм индукции микроядер представлял собой патологическую сегрегацию хромосом (анеугенность), но не кластогенное действие на хромосомы.
Снижение репродуктивной функции
Исследований по оценке эффекта препарата Алеценза на репродуктивную функцию на животных не проводились. В общих токсикологических исследованиях нежелательных эффектов на репродуктивные органы самцов и самок и не наблюдалось. Данные исследования проводились на крысах и обезьянах при экспозиции, соответственно в 2,6 и 0,5 или более раз превышающих экспозицию у человека, измерявшуюся в AUC, при применении в рекомендуемой дозе 600 мг два раза в сутки.
Тератогенность
Алектиниб вызывал токсическое воздействие на эмбрион и плод у беременных самок крыс и кроликов. У беременных крыс алектиниб вызывал потерю плода (выкидыш) при экспозиции, в 4,5 раз превышающей экспозицию у человека (на основании AUC), а также небольшой размер плода с задержкой формирования костей и незначительными патологиями органов при экспозиции, в 2,7 раз превышающей экспозицию у человека (на основании AUC). У беременных самок кроликов алектиниб вызывал потерю эмбриона или плода, небольшой размер плода и повышал частоту скелетных изменений при экспозиции, в 2,9 раз превышающей экспозицию у человека (на основании AUC) при приеме в рекомендуемой дозе.
Прочее
Алектиниб поглощает УФ-свет в диапазоне от 200 до 400 нм и демонстрировал наличие фототоксического потенциала в анализе фотобезопасности, проводившемся in vitro на культивированных фибробластах мыши после УФА-облучения.
В исследовании токсичности повторных доз целевыми органами, как у крыс, так и у обезьян при клинически значимой экспозиции были, среди прочего, эритроидная система, желудочно-кишечный тракт и гепатобилиарная система.
Патологическая морфология эритроцитов наблюдалась при экспозиции, в 10-60% превышающей экспозицию у человека (на основании AUC) при применении в рекомендуемых дозах. При экспозиции, в 20-120% превышающей экспозицию у человека (на основании AUC) при применении в рекомендуемых дозах, у обоих видов наблюдалось расширение пролиферативной зоны в слизистой ЖКТ. У крыс и/или обезьян при экспозиции, в 20-30% превышающей экспозицию у человека (на основании AUC) при применении в рекомендуемых дозах, повышался уровень печеночной щелочной фосфатазы (ЩФ) и прямого билирубина, а также наблюдалась вакуолизация/дегенерация/некроз эпителия желчных протоков и увеличение/очаговый некроз гепатоцитов.
У обезьян при клинически значимой экспозиции наблюдался слабый гипотензивный эффект.